![贵州吉创生物技术有限公司[官网]](/Public/Uploads/image/picture/160userid20180911_p1536645747.png)



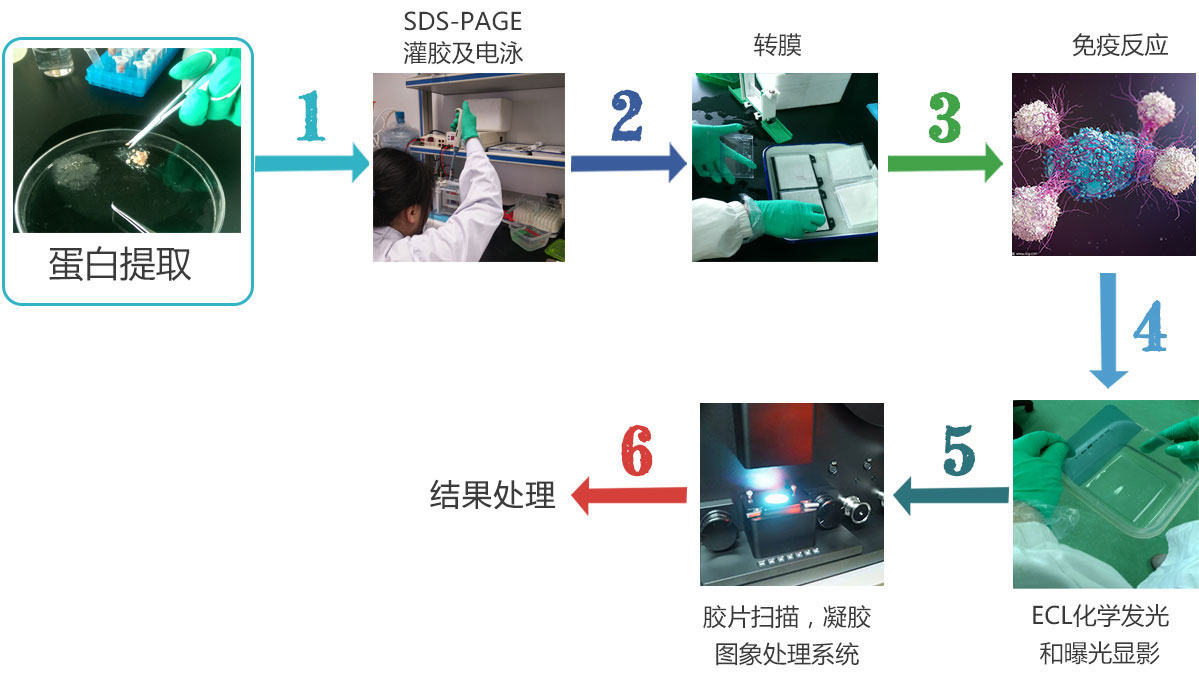

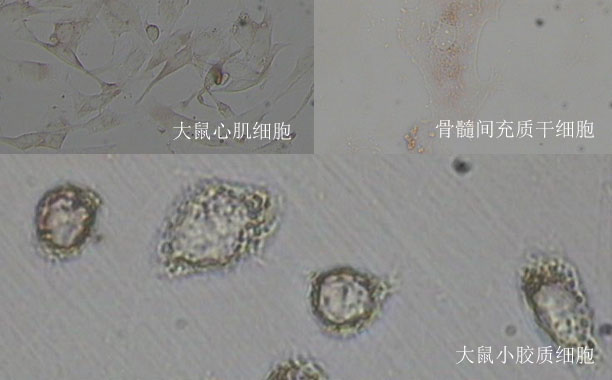





















WB实验指南
样品制备:
变性条件——SDS Loading Buffer直接裂解:
用常温或者高温预热过(预热更有利于阻止蛋白水解或者磷酸酶去磷酸化,但容易遗忘,LB久煮某些性质会改变)的5×SDS LB按比例加到细胞或者组织上并煮样。如果SDS LB不够,样品蛋白浓度过高时,煮样后会发现吸不上来,非常粘、一砣一砣的,很容易堵住枪头。这时候常规做法有两种,1. 再煮5min。常规煮样时间3-5min,样品过浓时就煮10min;2.如果10min煮样后,仍然吸不起来,才适当增加SDS LB,继续煮。煮样时间若过长,蛋白会凝固,此时以失去继续WB的意义,请丢弃(判断标准:出现明显的蛋白沉淀和水分层)
此方法的缺点,SDS LB煮过的样品如果用来做IP,需要特殊方法,因此,这种制备方法制备的样品有使用局限。
非变性裂解法(不讨论诸如核抽提、亚细胞器分离等等了,其实类似)
裂解细胞请尽量冰上操作,减缓酶解作用。
使用温和裂解液裂解(配方:20mM Tris/HCl, pH7.6, 100mM NaCl, 20mM KCl, 1.5mM MgCl2, 0.5% NP-40 and 0.5mM PMSF)。
此方法的优点:NaCl浓度略低于生理状态,保证裂解细胞的方式较为温和,便于随后的IP等试验。其他Na盐浓度的细胞裂解液也很常见,诸如0.15M(生理盐浓度)、0.3M、0.6M(高盐系列,裂解细胞时染色体会析出,细胞碎片和沉淀非常粘稠)及0.8-1.2M;0.3M及以下适合IP试验,0.6M及以上适合纯化蛋白,尤其相互作用较强的复合体,要得到纯化的一些复合体的组成蛋白一般采用极端的高盐裂解细胞。
组织块裂解:
组织块较大用匀浆的方法最合适,现在有不少转头很小的匀浆器。
当组织超微量的时候,比如50mg新鲜癌组织,我采用的方法是
1. 低温(剪)搅碎,成肉糜状。
2. 锡箔纸包裹好,放入少量液氮,快速敲击(控制力量,尽量避免弄破锡箔纸),进一步破碎;反复多次。
3. 用上述细胞裂解液回收。
样品制备完,应立即低温保存(-20度短期(几天);-80度长期;例外,IP用样品应直接进行IP,避免冻融破坏蛋白质间弱的相互作用)。SDS LB煮沸过的样品冻融会存在另一个问题,SDS沉淀;SDS 4度就会沉淀,何况-80度。上样前应加热充分溶解,否则从加样口向下拖带(SDS LB不够,样品未充分溶解也会出现类似拖尾;上样过大也会)。
在样品制备过程中,另一个需要注意的问题是,从样品制备的起始阶段就要注意定量问题;WB本身系统误差有20%,太细微的差别经常忽略不计。细胞样品可以先计数再接种,短时间内即使细胞生长有差异也不会对WB结果影响太多。组织样品,从肿瘤组织的大小(称重)开始,裂解液的体积都是精确定量的,操作过程也尽量避免蛋白损失。
蛋白电泳:
电泳胶的制备、配方、缓冲液配方,请参考分子克隆。经验之谈,8%胶最底边约36KDa,10%最底边约25KDa,12%最底部约12KDa。8%胶可以跑300KDa-36KDa之间的任意蛋白,转膜效率对WB结果的的影响都问题不大。12%,180KDa左右,转膜效率对WB结果的影响都问题不大(300KDa蛋白如何,没有尝试过)。
电泳一般采用恒流,45mA-55mA(应根据电泳仪器适当调整,要注意仪器最高限压;此条件适合gibco model V16型,胶宽20cm)。采用恒流的优点,保证最快速度完成电泳(电压会逐渐增大),省时且减少扩散;但是由于电泳速度过快时会发热而溶胶,所以你必须想办法散热。散热不好,条带出现波浪状。
注意事项:
1.聚丙烯酰胺的30%母液会降解,要4度避光保存。
2.APS会失效,10%APS一般保质期才一个月左右。-20度分装长期保存。
3.注意Tris buff的PH值,以及平时所用的水的PH值。PH值改变会使带型非常怪异,所有蛋白和溴酚蓝压成一条细线(即便在分离胶中),如果溴酚蓝前有红色染料,那么此染料彗星式拖尾从溴酚蓝一直延伸到胶底部(溴酚蓝此时涌动极缓慢,位于胶中上部)
4.上下层式电泳装置若漏液,哪边漏液,电泳条带往哪边倾斜。内外式电泳装置漏液,则装置处于短路状态,液体会过热。SDS-PAGE胶可能局部自溶,条带扭曲、变形。
5.配胶用玻璃板和边条应及时洗净。玻板未洗净的坏处很多。尽管玻璃看似平滑,但是一些细微的凹陷处会凝结肉眼无法分辨的胶颗粒(摸上去疙疙瘩瘩),其坏处是,在该部位极易导致不均匀的胶块,样品经过该处或电泳散热不好,条带变形。如果是RNA的超薄胶,胶板的颗粒会导致局部巨大气泡,非常难于清除;蛋白胶也会导致一些小气泡的产生。玻板没洗净的另一个坏处是,由中学物理知识可知,玻璃表面越光滑粘附越牢,未洗净的玻板会削弱和胶体间的粘附力,拔梳子或边条时会产生微小的错动,胶体下部出现大量气泡(夹在玻板和胶之间,这个关系不大);严重的错动会使上样时,样品从胶和玻璃之间的间隙漏光,或者甚至胶和玻板分开。为避免拔梳子时的错动,可在电泳缓冲液中拔梳子(比水更好,有SDS润滑)。
6.上样时,不要把枪头深入胶孔过深(或者采用细的尖端拉长的专用上样枪头),可能会错开胶和玻板,样品会泄露。
7.未加样的孔应添加高浓度SDS LB平衡,否则最外侧条带会拉宽变形。通常20μl样品(含4μl 5×SDS LB),可用8μl 5×SDS LB平衡。同理,点marker的lane也要加入同样体积的LB。LB若在室温放置太久和新鲜的在比重上会有差异,在新旧LB靠近的地方,条带会拉宽或挤压变形。因此,同一块胶上,煮样及填空白所用LB应一致。LB应-20度保存。
8.增加上样量不一定会提高荧光信号强度。增加上样量的后果通常只能是让你的内参粘连,所有的蛋白条带都扭曲变形。因为增加上样量最多只能提高几倍,而WB灵敏度是以10的几次幂量级的,所有目的蛋白信号的唯一方案就是IP富集(可特异提高目的蛋白浓度数百数千倍,并去除其它杂蛋白干扰)。增加上样量的另一个坏处是,本来高表达的蛋白,诸如内参,在同样WB条件下,可能出现荧光灼烧式粹灭(一晃而灭)或者条带中空。
仍以6孔板80%以上汇合度为例,细胞裂解液和SDS LB通常都是投入200μl(最少80μl,这样面积的培养板如果裂解液投入太少,回收时的损失就太大,上样就很难做到一致),而这种浓度条件下制备的样品,电泳时上样量要控制在5-6μl,最少2.5μl,最多10μl,10μl时内参基本已经开始粘连成一条线,带型出现波浪纹;当然并非不可以多上样,再多对WB结果影响不大(没有的仍然没有),且条带都很丑。
9. 不同样品上样时,可考虑将样品体积调成一致或类似。如果一组IP样品和一组细胞裂解液样品,前者通常洗脱体积为15μl,刚好+4μl 5×SDS LB达到常规20μl上样体积;后者第8点提到只上4μl,同样+4μl SDS LB(比重同,不会互相挤压),最好补加10μl DDW使总体积一致。通常这样不同条件获得的样品,中间最好用“空白”泳道隔开(空白仅指无蛋白,仍应加入8μl 5×SDS LB),这样才能确保每条带都很美观。
10. SDS PAGE胶配好暂时不用时,需用电泳缓冲液灌满空间(拔去梳子和底边边条后的间隙),用保鲜膜包裹防止液体蒸发,短期室温或稍长期4度保存。
转膜
很多时候新手会把WB结果无信号归咎于转膜不够充分,实际上转膜效率对最终结果的影响所占比重并不高,真正取决定性因素的是抗体识别能力的强弱,以及如何正确使用抗体,。
有一个简单的例证,Biorad提供的3mm厚滤纸初次使用时,通常转膜效果不理想(从预染的marker深浅可知),但对多数一般丰度的蛋白的检测没有任何影响(建议少用这种新滤纸做那种含量特别稀少或者转膜非常困难的蛋白)。没有很好的策略可以解决这个新滤纸的问题;尝试过浸泡(会泡烂的)、预电泳(会稍微好点)等等,效果均不理想。通常,最初一两次的使用就是用于转一般蛋白。每次用完后清水稍微冲洗一下表面盐分(要控制水流;水流太大,纸张会烂掉的),晾干再用;只要没冲烂可以一直反复使用。
其次,尽管转膜效率不起决定性作用,但由于WB的流程相对较长,所以每个步骤的叠加作用会被级数放大,因此在试验的每个步骤我们仍会力求更好。
转膜方法有半干转和湿转,这两种转印方法各有优劣。湿转适合所有的蛋白,转膜效率最佳,但靡费试剂、溶液,对普通分子量大小的蛋白转染操作时间长于半干转(下文会具体给出条件);半干转适合分子量较小的蛋白,省时、省试剂。蛋白分子大小如何界定呢?习惯上认为150KDa以上为大蛋白,其他均属于小蛋白,当然这只是一个相对标准。因此,通常对大蛋白的转印多数人会选择长时程的湿转,而且用半干转做大蛋白转印本来就是高手在悬崖上跳舞,此时转膜效率是否更高已经没有任何意义——我说过转印效率对WB结果不起决定因素。
湿转、半干转缓冲液配方请参考分子克隆,有必要指出的是,实际上用半干转缓冲液进行湿转效果也非常理想,通常这种缓冲液可以反复使用多次(≤5次常规1hr电转,如有3hrs长时程转染,缓冲液使用次数会减少),不过要注意初始电流(同样温度下,初始电流高,表明缓冲液中电解质剩余不多,为确保转膜温度,可开始或中途更换新鲜转膜液)。电转液中SDS增加蛋白的水溶性,促进蛋白在电转液中泳动。若不加SDS,蛋白会沉淀在胶上,但SDS过多会影响蛋白与膜的吸附。甲醇能使SDS与蛋白分离,甲醇浓度越高SDS与蛋白分离越快,但高浓度的甲醇对蛋白会有固定作用,不利于蛋白从胶里跑出来。一般来说甲醇的浓度为20%,但PVDF膜截留marker上交联的小分子颜料的能力很差,可考虑提升甲醇的浓度至25%(它对普通蛋白的转膜影响不太,颜料截留更多);大蛋白转印可考虑降低甲醇浓度,另外SDS必须添加。甲醇的另一个作用是降温,旧的电转液由于电解质的消耗和甲醇的挥发,电转时电流或电压变化更快、温度升高也更快,因此可考虑增加额外的甲醇;这点对半干转缓冲液的意义较大,因为半干转缓冲液消耗很慢,缓冲液放置的时间较久,甲醇挥发比较严重,可临时少量补加。
湿转一般采用稳压,电压控制在120-140V,时长1-3h(小蛋白控制在1h,大蛋白3h);半干转一般稳流,依millipore PVDF膜附带说明书,当2.5-3mA/cm2,电压25V(biorad半干仪额定值不能超过25V;amersham半干转仪,限压30V。通常跑不到这么大的电压,除非是新的滤纸或很久的转膜液,或常温放置的转膜液,或超大的胶),时间一般15--45min(常规用0.5h);如果是3mA/cm2,时间不要超过0.5。这两款仪器的限压要求,可能出于保护仪器的目的;amersham为防止过载,当电压>30V,~35V附近的时候,保险会自动跳掉、切断电转仪电源。至于UK的,即使整张大板胶室温电转3h以后,最大电压仍为18V(因此足见其散热性能的优良)。注明:以上半干转仪时间的要求是针对PVDF膜的,这里面有一个很有趣的问题。尽管厂家一再表明PVDF膜比NC膜对蛋白的截留更高(但NC带负电性,理论上它的吸附量应该更高,所以通常同样使用TBST这种低盐洗液背景比PVDF膜高),但PVDF膜对小分子的截留明显不如NC膜,因此交联在预染marker上的颜料小分子很容易穿过PVDF膜,造成转膜过度的假象(除可考虑提升甲醇浓度至25%外,也可以考虑增加marker的上样量);NC膜没有这种问题,所以通常它的转膜时间也是小蛋白1h、大蛋白3h,电流控制在200-300mA(16cm×9cm大胶300mA,如果胶面积小很多可以考虑降低,实际上相当于2.5-3mA/cm2左右)。NC膜唯一不如PVDF膜的地方,在我看来是它的强度:干燥的膜易碎。当然目前某些厂家为解决这个问题,将NC成分涂在另一种介质的表面,这种膜的支持强度和PVDF膜类似,但转膜效率稍差于纯NC膜,且有明显的正反面;因此制作转印三明治的时候要特别注意正反面。此外,20KDa以下的蛋白宜采用0.22μM孔径的转印膜,但也不是一定必须。
对于大蛋白转印,很多书本建议采用低浓度胶,如6% SDS-PAGE。但实际操作发现,6%太软,制作转印三明治的操作非常麻烦;且内参如Actin、tubulin都在45KDa附近,而6%胶底边溴酚蓝指示60KDa左右,除非采用坛子上有人提过的灌不同浓度的胶或者梯度胶,否则需要同时跑两块胶,因此也不是很推荐。个人经验认为300KDa以下8%胶(底边36KDa)都可以胜任,可以适当调整选择7-8%胶可以兼顾内参和大蛋白。
转膜最好在低温进行(高温会局部溶胶或降低转膜效率),尽管很多半干转仪没有具体要求,但用预冷过的电转液湿滤纸比未预冷的转印效率更高。因此,有条件建议湿转、半干转均在低温(4度)进行。此外,湿转缓冲液可以考虑转印前-20度预冷,时间不能长于2h,否则电泳液冻结。
制作转膜三明治的过程中,书中强调过滤纸、胶、膜的大小最好一致,否则没有胶、膜隔开部分的滤纸会因为直接接触而产生局部短路,降低内阻增加电(压)流,造成升温过快。但进口仪器随厂原包装附带的滤纸有限,如果每次都切割新滤纸,消耗过快、初次使用的膜转膜效率不高,且增加额外的操作。因此,实际上滤纸大一些也不太要紧,通常膜的大小会严格控制在与胶面积一致或略小一点点。操作时在保证每一层均无气泡的前提下,叠好后再碾压几个来回,力道要均匀、恰好;力量过大,胶会被拉伸,条带会扭曲、变形。碾压的目的不是为了驱赶气泡(完全可以做到叠加的时候每一层都无任何气泡;诸如采用多层薄滤纸时可以一张张的叠加),更重要的作用是让膜和胶贴得更紧密。可以理解的是,对于蛋白分子的大小而言,胶和膜之间的间距是数十万倍计的,因此更紧密的接触无疑是转膜成败的关键。
胶和膜需不需要泡呢?不少公司实验讲座时提出泡胶、泡膜,实际操作中没有发现泡和不泡的转膜效率有多大差别。实际上,胶只需要在电转液中浸一下即可(可以减少与滤纸或者膜之间的气泡);而膜也只要湿掉沾上电转液即可,同样多沾些缓冲液会有利于减少气泡(PVDF要先用甲醇湿润再投入缓冲液;NC直接进缓冲液)。
抗体孵育
——WB真正的难点:抗体的使用是一种艺术,一种抗体一个脾气。
对WB结果有决定性影响的因素是抗体的效价和如果正确使用手中的抗体。无论你如何提高自己的转膜技术,高效和低效之间往往只有数倍的差异,而抗体的效价可以差数千倍以上;同样,正确使用抗体可以保证抗体效价不被过分的拮抗,谋求特异性和非特异性背景之间差异的最大化。
封闭最短可以缩减到5min:
很多人把高背景或者黑板,认定为封闭不充分,其实这也是没有吃透WB(说实话,经常看到坛子上很多人这样给人答疑,非常的无语);实际上封闭(极端点)也是个可有可无的步骤,真正对降低背景起核心作用的是抗体稀释液中的组分。当你的抗体使用次数较多时,基本不用封闭也可以有较低的背景;如果抗体很新,其实封闭最短可以缩减到5min(没试过更短的,不一定不可以)。那什么导致高背景甚至黑板呢?抗体的质量不太好(主因),或者抗体稀释液的配方有问题(增大抗体稀释比略微有所改善)。封闭液的配方可以和抗体稀释液相同,也可以不同;通常封闭液是最简单(单方)的抗体稀释液,而抗体稀释液可能是复方。
封闭很少出状况。不过在脱脂奶做封闭剂的封闭液中,脱脂奶颗粒未完全溶解时,微小的颗粒会粘附在膜上增加星点状背景。
紧接封闭操作的是抗体孵育,之间不要用TBST漂洗。抗体孵育成败的关键首要在抗体本身的质量,包括抗体的效价和背景的强弱,然而历来商品化的抗体质量参差不齐。抗体公司在出售抗体的时候,在广告上会玩很多花样。A.不给整张膜标记的图示。很多抗体质量不好,背景高杂带多,如果靶蛋白区域较干净时,他们通常只给很窄的一条靶蛋白区域的图例。B.不给内源性蛋白标记的图示,用IP的样品取代。通过IP可以富集抗原,减少杂蛋白,通常很容易拿到背景很干净,信号很强的图例。C.用制备抗体时高表达的多肽做阳性样品,不给全长蛋白的图例。由于多肽可以IP富集,且不存在空间折叠的问题(通常裸露在外),因此很容易标记。除以上花样外,不少抗体用途上还有局限,因此挑选抗体时要睁大眼睛。
一抗通常4度过夜,效率较常温1-1.5h高;二抗通常1:5000稀释,室温0.5-1h(过久并不会明显提高信号强度甚至会减弱(相当于洗膜),同时造成高背景进一步影响特异与非特异荧光信号强度间的对比。孵育时间过久还会迫使你延长TBST的时间,这对抗原识别能力较弱的抗体更致命)。洗膜通常用TBST,也可以考虑高盐洗膜液(NaCl 0.5M);洗膜时间一般3×10min或者4×5min。抗体孵育和洗膜时,摇床速度不能过快也不宜过慢,需要简单摸索一下。
如何通过改变抗体稀释液的配方,寻求抗体孵育的最佳条件呢?
首先要认清,抗体稀释液没有一个绝对通用的配方;一种抗体一个脾气。
其次,抗体稀释液的配方要兼顾与特异性抗体竞争抗原的强度,以及对其他区域的封闭强度两方面的平衡。
目前,抗体稀释液中可以充当封闭蛋白的主要有milk,BSA和gelatin,似乎很少的样子。。。可是,你有没有考虑过“复方”?——这下配方无限多了吧。
1. 采用milk,BSA和gelatin的单方。3% milk,5% BSA,<=0.4%的gelatin,并根据实际情况改变(若出现白板,请降低封闭剂浓度。;黑板多加)。gelatin大家可能比较陌生,不过如果你翻翻抗体公司卖给你的原装抗体里面液体的配方,你会发现很多抗体溶液里都有它。
2.很多情况下单方的浓度很高了,背景问题仍不能解决。那么请尝试两两或者三种不同比例混合。不过有些细节还是要自己摸索的,比如gelatin+BSA时,gelatin浓度不能无限提高(会完全竞争掉抗原抗体特异性结合,导致白板),BSA的浓度较随意。
3.抗体稀释液可用低盐浓度的TBST,也可用高盐浓度的缓冲液(NaCl 0.5M)以降低背景;但是切记,有的抗体对盐浓度敏感,包括构象和溶解度,因此需要尝试。
如果以上策略仍然无法解决高背景,可将稀释好的一抗先跟平时实验剪切下来的membrane的边角料(新的)放在一起孵育个把小时再用。如果还不行,彻底无解,结论抗体太差;请更换别的公司或者抗别的区段的同种抗体。
这三种封闭蛋白的差异:
milk和gelatin封闭效果不错且价格便宜,但milk容易发霉变质,且国产milk脱脂不充分亦增加背景、拮抗抗原抗体结合(拮抗与抗原结合尤其突出;更不知道三聚氰胺会不会影响抗原抗体结合或者ECL!?);BSA封闭效果不佳,且价格较昂贵。个人推荐gelatin,gelatin主要成分是骨胶原,蛋白很大,但可以通过加热打断成小蛋白,从而实现封闭各种分子量大小的蛋白。通常,我会将gelatin加到TBST中后直接灭菌,灭菌过程中,不溶的gelatin会逐渐溶解;由于gelatin可以灭菌(milk不可以),所以同样条件下,gelatin的存放时间明显长于milk。实际上,抗体可以反复用很久(通常一般的抗体常规稀释比,可以连续用一个月以上;对于老手用过2、3次的旧抗体更prefer,因此背景降到很低而效价正是最高的时候),通常抗体最后失效的原因不是因为使用次数过多,而是染菌;比较旧的抗体都会有很多的沉淀在底部,更长时间还会有异味。稀释的一抗靠添加NaN3并于4度保存延长使用时间;二抗不能加NaN3,但放在-20度反复冻融延长使用寿命(抗体并非绝对不可以冻融;只是高浓度的原始管抗体的冻融对效价会有较大影响)。
抗体稀释液(一抗、二抗稀释液也可同也可不同)和封闭液可以相同,可以不同,因为抗体稀释液可以采用复方的,所以即使混合了少许,不会对抗体的使用产生较大的影响。不过封闭液用milk时,gelatin稀释的一抗混入少许milk会影响其使用寿命。
由于以上诸多可变因素,实在无法总结出一种万金油式的抗体稀释液(只能是多数通用),因此对待具体问题时,请具体对待(多花点心思摸索吧)。
原装抗体的保存:
1. 分装。每次一管,但是可能太多占地方,而且还会有粘壁的损失,不是很推荐。
2. 1:1加甘油,冻于-20度,可长期保存,此方法实际上也被很多国内的试剂公司广泛采用,诸如博士得,中杉,它们小包装的进口抗体通常都是1:1添加甘油保存于-20(不信去查查)。值得注意的是:不是所有的抗体都可以1:1添加甘油,要以抗体说明书为准,如果说明书中本身存在甘油,或特殊注明外,通常可以采用。
显影
——淬灭的祸因
当抗体孵育、TBST漂洗结束后,进入ECL显影步骤。当然,目前国内一些实验室已经换用仪器扫描荧光信号,而我们更是二抗直接连荧光蛋白连ECL和常规的底片显影都省略了,因此以下所讨论的某些细节问题可能不再出现。
首先坛子上还有少数实验室处于DAB显色时代,奉劝一句,都走到最后一步了就不要节省了;DAB显色的灵敏性是远远不够的。目前较为流行的仍是化学发光法,下面简述一下其原理:
交联在二抗上的HRP酶能够特异水解过氧化物产生化学能;ECL中主要包含两种成分,催化剂和中间递质(白色瓶子主要是过氧化物如双氧水)。催化剂的作用不用废话了,其中间递质主要是吸收化学能并将其传递到luminol之类的化合物上产生电子跃迁从而激发荧光。因此催化剂的效率和中间递质的传递效率直接影响荧光的强弱。本人曾自制过ECL并尝试改良,其间发现很多化合物、金属离子如Fe、Cu等能催化并高效传递能量,且不依赖于HRP浓度,荧光信号能瞬间曝强;这其实与下文提到的一些粹灭原因有联系。
当初我自制的ECL最大的毛病在于稳定性不好,同理,商品化的ECL同样也有保质期的问题,往往最先失效的组分就是过氧化物,尽管他们必然添加过抗还原剂。其实,检验ECL是否失效的方法很简单,取稀释的即用型二抗1-2ul加至ECL混和液中观察荧光强度,即可知道ECL是否失效。
商品化的ECL价格并不便宜,因此ECL孵育的最佳方案是:将ECL滴加在保鲜膜上或干净的支持物上,如废弃的旧底片、塑料盒,有机玻璃盒等等(注意,国产的保鲜膜很多质量不过关,有两种潜在的问题,下面详述),然后倒扣膜于保鲜膜上,夹住一个角来回拖动数次至ECL均匀(有的产品说明书要求孵育1min)。另外平铺一张干净的保鲜膜,将膜揭下控干ECL、倒扣包好,然后显影。mini3型胶整张膜孵育,ECL总体积0.7-1ml足矣。
哪些因素会影响荧光信号的强弱呢?(这部分实际上与导致荧光淬灭的因素部分重合)
1.保鲜膜的质量。a.国产的保鲜膜,很多厂家偷工减料打价格战,造成保鲜膜很薄亦破损。ECL滴加到保鲜膜上容易从肉眼不易分辩的小孔泄露,一方面ECL反应不充分,另一方面ECL所含液体会溶解试验台上残留未知的化学药品颗粒(也许你或其他人曾经不小心打翻过某种化合物)、灰尘等等,造成荧光淬灭。在简述原理时,我曾经提到过很多化合物、金属离子能直接催化过氧化物水解,在极短的时间内消耗掉所有的HRP酶,荧光转瞬即逝。
b. 保鲜膜的化工原料或拉制过程中,残留未知的化学试剂,引起荧光淬灭;廉价的保鲜膜问题尤多。
目前,国产的就“妙洁”比较过关,保鲜膜厚度和化学物残留都不影响ECL显影。
如果买不到合格的保鲜膜,也可以用其他物品替代;但要特别注意这些支持物表面的干净,最好不要有任何化学试剂残留,包括风干的TBST盐结晶颗粒。
2.ECL孵育结束后膜需要控干。中学物理学过水会吸收光谱,因此任何液体包括ECL溶液本身都会干扰荧光强度,所以ECL孵育结束时需要控干。一般夹起膜,让膜的一角或一边接触吸水纸;但是请注意不能让膜完全干掉。某些信号较弱的ECL,膜彻底干掉后荧光会消失;较好的试剂盒,荧光相对稳定,但完全吸干以后,背景荧光也会升高,而且会严重影响重新标记新抗体时的背景。因此,按照我的方法,让自由流下的多余的ECL被吸走就好。
3.控干的膜要仔细用保鲜膜包被,防止接触外界异物造成荧光淬灭;同时,包裹保鲜膜时要细心,尽量不让正面保鲜膜和膜之间出现皱褶。皱褶的地方会堆积多余的ECL,要么形成一条荧光的亮线;要么由于液体吸收荧光或者局部区域HRP过度消耗,呈线条状淬灭。
4.在膜放入压片夹之前,最好肉眼观察一下荧光信号的强弱,这有利于判断实验可能出现的问题。很多人提问看见很强的荧光了,但怎么压片都没有信号,那么就是快速淬灭(闪灭,再怎么快速操作也拿不到这种情况下的荧光信号)造成的;有这个观察至少能够判断ECL孵育之前实验操作应该问题不大,而问题主要是淬灭。如果你省略这个步骤,那问题就非常复杂了,因为之前任何操作都可能导致白板,根本无法判断。
除上述的问题以外,还有哪些因素会导致荧光淬灭呢?
1. 抗原浓度太高,荧光信号过强,快速淬灭。在电泳的章节就曾提过,如果上样量太大,不仅条带会粘连、弯曲,更可能导致淬灭。因为局部区域过多的HRP酶会快速消耗底物呈富氧态,条带出现烧灼样(取出膜会发现一条明显的暗黄色的带),荧光信号会闪灭或者条带空心(条带灰黑不实,则可能是显影液接近失效)。
2. 抗原浓度太低,荧光信号太弱,相对慢一点的淬灭。可能第一次压片能够获得很弱的条带,随后都不可见。
3. ECL试剂质量。除过氧化物因接触空气被逐渐还原外,ECL的成分通常都不稳定。如呈递电子跃迁能的中间递质其化学性质就不太稳定,因为它转换能量的特性决定其必是一个有中间态的化合物,长期接触空气会被逐渐氧化失效;luminol也有保质期。
4. 操作时间过长,膜逐渐彻底干掉。商品化的ECL会出现波形渐变,开始信号逐渐增强,背景一起升高;随后荧光完全衰减淬灭。
本文链接: http://www.jcswbio.com/news/8041.html